I am pleased to announce that the next phase of BioJulia is starting! In the next several months, I'm going to implement many crucial features for bioinformatics that will motivate you to use Julia and BioJulia libraries in your work. But before going to the details of the project, let me briefly introduce you what the BioJulia project is. This project is supported by the Moore Foundation and the Julia project.
The BioJulia project is a collaborative open source project to create an infrastructure for bioinformatics in the Julia programming language. It aims to provide fast and accessible software libraries. Julia's Just-In-Time (JIT) compiler enables this greedy goal without resorting to other compiled languages like C/C++. The central package developed under the project is Bio.jl, which provides fundamental features including biological symbols/sequences, file format parsers, alignment algorithms, wrappers for external softwares, etc. It also supports several common file formats such as FASTA, FASTQ, BED, PDB, and so on. Last year I made the FMIndexes.jl package to build a full-text search index for large genomes as a Julia Summer of Code (JSoC) student, and we released the first development version of Bio.jl. While the BioJulia project is getting more active and the number of contributors are growing, we still lack some important features for realistic applications. Filling in gaps between our current libraries and actual use cases is the purpose of my new project.
So, what will be added in it? Here is the summary of my plan:
Sequence analysis:
Online sequence search algorithms
Data structure for reference genomes
Error-correcting algorithms for DNA barcodes
Parsers for BAM and CRAM file formats
Integration with data viewers and databases:
Genome browser backend
Parsers for GFF3 and VCF/BCF
Database access through web APIs
These things are of crucial importance for writing analysis programs because they connects software components (e.g. programs, archives, databases, viewers, etc.); data analysis softwares in bioinformatics usually read/write formatted data from/to each other. The figure below shows common workflow of detecting genetic variants; underlined deliverables will connect softwares, archives and databases so that you can write your analysis software in the Julia language.
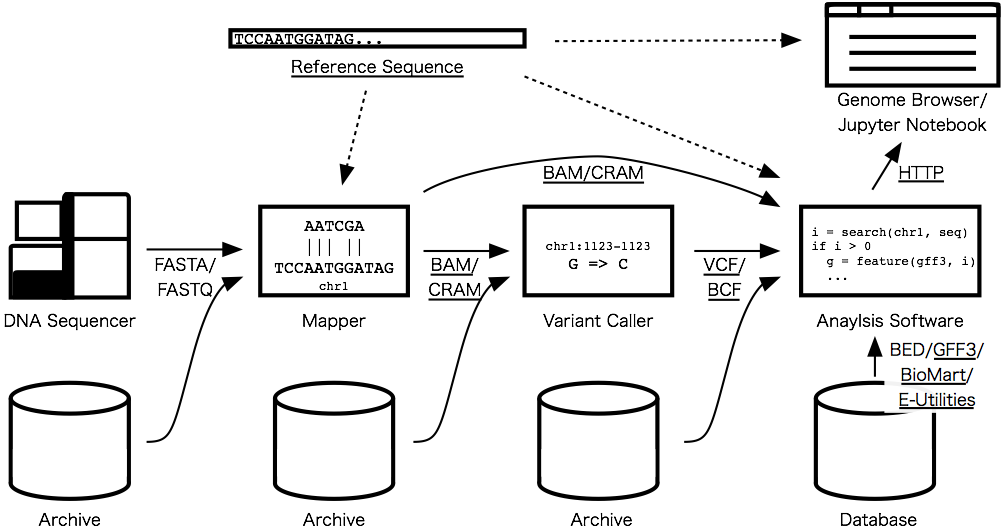
Sequence Analysis
The online sequence search algorithms will come with three flavors: exact, approximate, and regular expression search algorithms. The exact sequence search literally means finding exactly matching positions of a query sequence in another sequence. The approximate search is similar to the exact search but allows up to a specified number of errors: mismatches, insertions, and deletions. The regular expression search accepts a query in regular expression, which enables flexible description of a query pattern like motifs. For these algorithms, there are already half-done pull requests I'm working on: #152, #153, #143.
After the last release of Bio.jl v0.1.0, the sequence data structure has been significantly rewritten to make biological sequence types coherent and extensible. But because we chose an encoding that requires 4 bits per base to represent DNA sequences, the DNA sequence type consumes too much memory than necessary to store a reference genome, which is usually composed of four kinds of DNA nucleotides (denoted by A/C/G/T) and (consecutive and relatively small number of) undetermined nucleotides (denoted by N). After trying some data structures, I found that memory space of N positions can be dramatically saved using IndexableBitVectors.jl, which is a package I created in JSoC 2015. I'm developing a separated package for reference genomes, ReferenceSequences.jl, and going to improve the functionality and performance to handle huge genomes like the human genome.
If you are a researcher or an engineer who handles high-throughput sequencing data, BAM and CRAM parsers would be the most longing feature addition in the list. BAM is the de facto standard file format to accommodate aligned sequences and most sequence mappers generate alignments in this format. CRAM is a storage-efficient alternative of BAM and is getting popular reflecting explosion of accumulated sequence data. Since these files contain massive amounts of DNA sequences from high-throughput sequencing machines, high-speed parsing is a practically desirable feature. I'm going to concentrate on the speed by careful tuning and multi-thread parallel computation which is planned to be introduced in the next Julia release.
Integration with Data Viewers and Databases
Genome browsers enable to interactively visualize genetic features found in individuals and/or populations. For example, using the UCSC Genome Browser, you can investigate genetic regions along with sequence annotations around the ABO gene in a window. Genome browser is one of the most common visualizations and hence lots of softwares have been developed but unfortunately there is no standardized interface. So, we will need to select a promising one that is an open source and supporting interactions with other softwares. The first candidate is JBrowse, which is built with modern JavaScript and HTML5 technologies. It also supports RESTful APIs and hence it can fetch data from a backend server via HTTP. I'm planning to make an API server that responds to queries from a genome browser to interactively visualize in-memory data.
Many databases distribute their data in some standardized file formats. As for genetic annotations and variants, GFF3 and VCF would be the most common formats. If you are using data from human or mouse, you should know various annotations are available from the GENCODE project. It offers data in GTF or GFF3 file formats. NCBI provides human variation sets in VCF file formats here. These file formats are text, so you may think it is trivial to write parsers when you need them. It is partially true — if you don't care about completeness and performance. Parsing a text file format in a naive way (for example, split a line by a tab character) allocates many temporary objects and often leads to degrade performance, while careful tuning of a parser leads to complicated code that is hard to maintain. @dcjones challenged this problem and made a great work and made Julia support for Ragel, which generates Julia code that executes a finite state machine. Daniel's talk of the JuliaCon 2015 is helpful to know about the details if you are interested:
Sometimes you may need only a part of data provided by a database. In such a case, web-based APIs are handy to fetch necessary data on demand. BioMart Central Portal offers a unified access point to a range of biological databases that is programmatically accessible via REST and SOAP APIs. Julian wrapper to BioMart will make it much easier to access data by automatically converting response to Julia objects. In the R language, the biomaRt package is one of the most downloaded packages in Bioconductor packages: https://www.bioconductor.org/packages/stats/.
Try BioJulia!
We need users and collaborators of our libraries. Feedbacks from users in the real world are the most precious thing to improve the quality of our libraries. We welcome feature requests and discussions that will make bioinformatics easier and faster. Tools for phylogenetics and structural biology, which I didn't mention in this post, are also under active development. You can post issues here: https://github.com/BioJulia/Bio.jl/issues; if you want to get in touch with us more casually, this Gitter room may be more convenient: https://gitter.im/BioJulia/Bio.jl.